|
|
| Line 162: |
Line 162: |
| | ====DESMOND==== | | ====DESMOND==== |
| | ====GROMACS==== | | ====GROMACS==== |
| − | Download and general information: http://www.gromacs.org
| + | Please refer to the [[gromacs|GROMACS]] page |
| − | | |
| − | Search the mailing list archives: http://oldwww.gromacs.org/swish-e/search/search2.php
| |
| − | | |
| − | =====Peculiarities of running single node GROMACS jobs on SCINET=====
| |
| − | This is '''VERY IMPORTANT !!!'''
| |
| − | Please read the [[https://support.scinet.utoronto.ca/wiki/index.php/User_Tips#Running_single_node_MPI_jobs relevant user tips section]] for information that is essential for your single node (up to 8 core) MPI GROMACS jobs. | |
| − | | |
| − | -- [[User:Cneale|cneale]] 14 September 2009
| |
| − | | |
| − | =====Compiling and Running GROMACS on Scinet (general information)=====
| |
| − | Chris Neale has compiled gromacs on GPC, with assistance from Scott Northrup, and
| |
| − | on the power6 cluster with assistance from Ching-Hsing Yu. Users are welcome to utilize
| |
| − | these binary executables, but only at their own peril since compiling and testing your own
| |
| − | executable is safer and more stable.
| |
| − | | |
| − | ''Gromacs executables:''
| |
| − | | |
| − | '''GPC:''' /scratch/cneale/exe/intel/gromacs-4.0.5/exec/bin
| |
| − | | |
| − | '''TCS:''' /scratch/cneale/exe/gromacs-4.0.4_aix/exec/bin
| |
| − | | |
| − | Below you will find, in order, scripts for the different compilations that you can follow
| |
| − | to make your own binaries.
| |
| − | | |
| − | '''NOTE:''' ''the steps are not listed in order! You must compile fftw before compiling gromacs, and if you are going to use mvapich2-1.4rc1 then you must compile it also before compiling parallel gromacs.''
| |
| − | | |
| − | # Compiling serial single precision gromacs on GPC
| |
| − | # Compiling openmpi parallel gromacs on GPC
| |
| − | # Compiling serial gromacs on the power6 (submitted to the queue):
| |
| − | # Compiling parallel gromacs on the power6 (submitted to the queue):
| |
| − | # fftw single precision compilation
| |
| − | # Change to get mvapich2-1.4rc1 to compile gromacs
| |
| − | # Compiling mvapich2-1.4rc1
| |
| − | # Compiling gromacs on GPC using mvapich2-1.4rc1
| |
| − | # Submitting an IB GPC job using openmpi
| |
| − | # Submitting an IB GPC job using mvapich2-1.4rc1
| |
| − | # Submitting a non-IB GPC job using openmpi
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | | |
| − | =====Compiling serial single precision gromacs on GPC=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | cd /scratch/cneale/exe/intel/gromacs-4.0.5
| |
| − | mkdir exec
| |
| − | module purge
| |
| − | module load intel
| |
| − | export FFTW_LOCATION=/scratch/cneale/exe/intel/fftw-3.1.2/exec
| |
| − | export GROMACS_LOCATION=/scratch/cneale/exe/intel/gromacs-4.0.5/exec
| |
| − | export CPPFLAGS=-I$FFTW_LOCATION/include
| |
| − | export LDFLAGS=-L$FFTW_LOCATION/lib
| |
| − | ./configure --prefix=$GROMACS_LOCATION --without-motif-includes
| |
| − | --without-motif-libraries --without-x --without-xml >output.configure
| |
| − | 2>&1
| |
| − | make >output.make 2>&1
| |
| − | make install >output.make_install 2>&1
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Compiling openmpi parallel gromacs on GPC:=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | cd /scratch/cneale/exe/intel/gromacs-4.0.5
| |
| − | mkdir exec
| |
| − | module purge
| |
| − | module load openmpi intel
| |
| − | export FFTW_LOCATION=/scratch/cneale/exe/intel/fftw-3.1.2/exec
| |
| − | export GROMACS_LOCATION=/scratch/cneale/exe/intel/gromacs-4.0.5/exec
| |
| − | export CPPFLAGS="-I$FFTW_LOCATION/include
| |
| − | -I/scinet/gpc/mpi/openmpi/1.3.2-intel-v11.0-ofed/include
| |
| − | -I/scinet/gpc/mpi/openmpi/1.3.2-intel-v11.0-ofed/lib"
| |
| − | export LDFLAGS=-L$FFTW_LOCATION/lib
| |
| − | /gpc/mpi/openmpi/1.3.2-intel-v11.0-ofed/lib/openmpi
| |
| − | -I/scinet/gpc/x1/intel/Compiler/11.0/081/lib/intel64
| |
| − | -I/scinet/gpc/x1/intel/Compiler/11.0/081/mkl/lib/em64t/"
| |
| − | ./configure --prefix=$GROMACS_LOCATION --without-motif-includes
| |
| − | --without-motif-libraries --without-x --without-xml --enable-mpi
| |
| − | --program-suffix="_openmpi" >output.configure.mpi 2>&1
| |
| − | make >output.make.mpi 2>&1
| |
| − | make install-mdrun >output.make_install.mpi 2>&1
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Compiling serial gromacs on the power6 (submitted to the queue)=====
| |
| − | | |
| − | Note that the -O5 flag for the power6 compilation makes it take about
| |
| − | 20 hours to compile. You can drop that if you want, but it does give
| |
| − | you a few more percent performance.
| |
| − | | |
| − | <source lang="sh">
| |
| − | #======================================================================
| |
| − | # Specifies the name of the shell to use for the job
| |
| − | # @ shell = /usr/bin/ksh
| |
| − | # @ job_type = serial
| |
| − | # @ class = verylong
| |
| − | ## # @ node = 1
| |
| − | ## # @ tasks_per_node = 1
| |
| − | # @ output = $(jobid).out
| |
| − | # @ error = $(jobid).err
| |
| − | # @ wall_clock_limit = 40:00:00
| |
| − | #=====================================
| |
| − | ## this is necessary in order to avoid core dumps for batch files
| |
| − | ## which can cause the system to be overloaded
| |
| − | # ulimits
| |
| − | # @ core_limit = 0
| |
| − | #=====================================
| |
| − | ## necessary to force use of infiniband network for MPI traffic
| |
| − | ### TURN IT OFF # @ network.MPI = csss,not_shared,US,HIGH
| |
| − | #=====================================
| |
| − | # @ environment=COPY_ALL
| |
| − | # @ queue
| |
| − | export
| |
| − | PATH=/usr/lpp/ppe.hpct/bin:/usr/vacpp/bin:.:/usr/bin:/etc:/usr/sbin:/usr/ucb:/usr/bin/X11:/sbin:/usr/java14/jre/bin:/usr/java14/bin:/usr/lpp/LoadL/full/bin:/usr/local/bin
| |
| − | export F77=xlf_r
| |
| − | export CC=xlc_r
| |
| − | export CXX=xlc++_r
| |
| − | export FFLAGS="-O5 -qarch=pwr6 -qtune=pwr6"
| |
| − | export CFLAGS="-O5 -qarch=pwr6 -qtune=pwr6"
| |
| − | export CXXFLAGS="-O5 -qarch=pwr6 -qtune=pwr6"
| |
| − | export FFTW_LOCATION=/scratch/cneale/exe/fftw-3.1.2_aix/exec
| |
| − | export GROMACS_LOCATION=/scratch/cneale/exe/gromacs-4.0.4_aix/exec
| |
| − | export CPPFLAGS=-I$FFTW_LOCATION/include
| |
| − | export LDFLAGS=-L$FFTW_LOCATION/lib
| |
| − | cd /scratch/cneale/exe/gromacs-4.0.4_aix
| |
| − | mkdir exec
| |
| − | ./configure --prefix=$GROMACS_LOCATION --without-motif-includes
| |
| − | --without-motif-libraries --without-x --without-xml >output.configure
| |
| − | 2>&1
| |
| − | make >output.make 2>&1
| |
| − | make install >output.make_install 2>&1
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Compiling parallel gromacs on the power6 (submitted to the queue)=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | #===============================================================================
| |
| − | # Specifies the name of the shell to use for the job
| |
| − | # @ shell = /usr/bin/ksh
| |
| − | ##### @ job_type = serial
| |
| − | # @ job_type = parallel
| |
| − | # @ class = verylong
| |
| − | # @ node = 1
| |
| − | # @ tasks_per_node = 1
| |
| − | # @ output = $(jobid).out
| |
| − | # @ error = $(jobid).err
| |
| − | # @ wall_clock_limit = 40:00:00
| |
| − | #=====================================
| |
| − | ## this is necessary in order to avoid core dumps for batch files
| |
| − | ## which can cause the system to be overloaded
| |
| − | # ulimits
| |
| − | # @ core_limit = 0
| |
| − | #=====================================
| |
| − | ## necessary to force use of infiniband network for MPI traffic
| |
| − | ### TURN IT OFF # @ network.MPI = csss,not_shared,US,HIGH
| |
| − | #=====================================
| |
| − | # @ environment=COPY_ALL
| |
| − | # @ queue
| |
| − | | |
| − | export F77=xlf_r
| |
| − | export CC=xlc_r
| |
| − | export CXX=xlc++_r
| |
| − | export FFLAGS="-O5 -qarch=pwr6 -qtune=pwr6"
| |
| − | export CFLAGS="-O5 -qarch=pwr6 -qtune=pwr6"
| |
| − | export CXXFLAGS="-O5 -qarch=pwr6 -qtune=pwr6"
| |
| − | export FFTW_LOCATION=/scratch/cneale/exe/fftw-3.1.2_aix/exec
| |
| − | export GROMACS_LOCATION=/scratch/cneale/exe/gromacs-4.0.4_aix/exec
| |
| − | export CPPFLAGS=-I$FFTW_LOCATION/include
| |
| − | export LDFLAGS=-L$FFTW_LOCATION/lib
| |
| − | cd /scratch/cneale/exe/gromacs-4.0.4_aix
| |
| − | echo "cn-r0-10" > ~/.rhosts
| |
| − | echo localhost > ~/host.list
| |
| − | for((i=2;i<=16;i++)); do
| |
| − | echo localhost >> ~/host.list
| |
| − | done
| |
| − | export MP_HOSTFILE=~/host.list
| |
| − | ./configure --prefix=$GROMACS_LOCATION --without-motif-includes
| |
| − | --without-motif-libraries --without-x --without-xml --enable-mpi
| |
| − | --disable-nice --program-suffix="_mpi" CC=mpcc_r F77=mpxlf_r >
| |
| − | output.configure_mpi 2>&1
| |
| − | make mdrun > output.make_mpi 2>&1
| |
| − | make install-mdrun > output.make_install_mpi 2>&1
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====fftw single precision compilation=====
| |
| − | | |
| − | FFTW is required by GROMACS. This compilation must be completed before compiling GROMACS.
| |
| − | | |
| − | <source lang="sh">
| |
| − | mkdir exec
| |
| − | export FFTW_LOCATION=/scratch/cneale/exe/intel/fftw-3.1.2/exec
| |
| − | module purge
| |
| − | module load openmpi intel
| |
| − | ./configure --enable-float --enable-threads --prefix=${FFTW_LOCATION}
| |
| − | make
| |
| − | make install
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Change to get mvapich2-1.4rc1 to compile gromacs=====
| |
| − | | |
| − | This change is required to the mvapich2-1.4rc1 source code in order to compile GROMACS with it.
| |
| − | | |
| − | <source lang="sh">
| |
| − | src/mpid/ch3/channels/mrail/src/gen2/ibv_channel_manager.c
| |
| − | line 503
| |
| − | unsigned long debug = 0;
| |
| − | to
| |
| − | static unsigned long debug = 0;
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Compiling mvapich2-1.4rc1=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | cd /scratch/cneale/exe/mvapich2-1.4rc1
| |
| − | mkdir exec
| |
| − | module purge
| |
| − | module load intel
| |
| − | ./configure --prefix=/scratch/cneale/exe/mvapich2-1.4rc1/exec CC=icc
| |
| − | CXX=icpc F90=ifort F77=ifort >output.configure 2>&1
| |
| − | make >output.make 2>&1
| |
| − | make install >output.make_install 2>&1
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Compiling gromacs on GPC using mvapich2-1.4rc1=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | #!/bin/bash
| |
| − | cd /scratch/cneale/exe/intel/gromacs-4.0.5
| |
| − | mkdir exec
| |
| − | PATH=/usr/lib64/qt-3.3/bin:/usr/kerberos/sbin:/usr/kerberos/bin:/usr/local/sbin:/usr/local/bin:/sbin:/bin:/usr/sbin:/usr/bin:/opt/xcat/bin:/opt/xcat/sbin:/root/bin:/opt/torque/bin:/opt/xcat/bin:/opt/xcat/sbin:/usr/lpp/mmfs/bin:/scratch/cneale/exe/mvapich2-1.4rc1/exec/bin/:/scinet/gpc/x1/intel/Compiler/11.0/081/bin/intel64
| |
| − | LD_LIBRARY_PATH=/scratch/cneale/exe/mvapich2-1.4rc1/exec/lib/:/scinet/gpc/x1/intel/Compiler/11.0/081/lib/intel64:/scinet/gpc/x1/intel/Compiler/11.0/081/mkl/lib/em64t/
| |
| − | export FFTW_LOCATION=/scratch/cneale/exe/intel/fftw-3.1.2/exec
| |
| − | export GROMACS_LOCATION=/scratch/cneale/exe/intel/gromacs-4.0.5/exec
| |
| − | export CPPFLAGS="-I$FFTW_LOCATION/include
| |
| − | -I/scratch/cneale/exe/mvapich2-1.4rc1/exec/include
| |
| − | -I/scratch/cneale/exe/mvapich2-1.4rc1/exec/lib"
| |
| − | export LDFLAGS=-L$FFTW_LOCATION/lib
| |
| − | ./configure --prefix=$GROMACS_LOCATION --without-motif-includes
| |
| − | --without-motif-libraries --without-x --without-xml --enable-mpi
| |
| − | --program-suffix="_mvapich2" >output.configure.mpi.mvapich2 2>&1
| |
| − | make >output.make.mpi.mvapich2 2>&1
| |
| − | make install-mdrun >output.make_install.mpi.mvapich2 2>&1
| |
| − | make distclean
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Submitting an IB GPC job using openmpi=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | #!/bin/bash
| |
| − | #PBS -l nodes=10:ib:ppn=8,walltime=40:00:00,os=centos53computeA
| |
| − | #PBS -N 1
| |
| − | if [ "$PBS_ENVIRONMENT" != "PBS_INTERACTIVE" ]; then
| |
| − | if [ -n "$PBS_O_WORKDIR" ]; then
| |
| − | cd $PBS_O_WORKDIR
| |
| − | fi
| |
| − | fi
| |
| − | /scinet/gpc/mpi/openmpi/1.3.2-intel-v11.0-ofed/bin/mpirun -np $(wc -l
| |
| − | $PBS_NODEFILE | gawk '{print $1}') -machinefile $PBS_NODEFILE
| |
| − | /scratch/cneale/exe/intel/gromacs-4.0.5/exec/bin/mdrun_openmpi -deffnm
| |
| − | pagp -nosum -dlb yes -npme 24 -cpt 120
| |
| − | ## To submit type: qsub this.sh
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Submitting an IB GPC job using mvapich2-1.4rc1=====
| |
| − | | |
| − | Note that mvapich2-1.4rc1 is not configured to fall back to ethernet
| |
| − | so this will not work on the non-IB nodes, even for 8 cores.
| |
| − | | |
| − | <source lang="sh">
| |
| − | #!/bin/bash
| |
| − | #PBS -l nodes=4:ib:ppn=8,walltime=30:00:00,os=centos53computeA
| |
| − | #PBS -N 1
| |
| − | if [ "$PBS_ENVIRONMENT" != "PBS_INTERACTIVE" ]; then
| |
| − | if [ -n "$PBS_O_WORKDIR" ]; then
| |
| − | cd $PBS_O_WORKDIR
| |
| − | fi
| |
| − | fi
| |
| − | module purge
| |
| − | module load mvapich2 intel
| |
| − | /scratch/cneale/exe/mvapich2-1.4rc1/bin/mpirun_rsh -np $(wc -l
| |
| − | $PBS_NODEFILE | gawk '{print $1}') -hostfile $PBS_NODEFILE
| |
| − | /scratch/cneale/exe/intel/gromacs-4.0.5/exec/bin/mdrun_mvapich2
| |
| − | -deffnm pagp -nosum -dlb yes -npme 12 -cpt 120
| |
| − | ## To submit type: qsub this.sh
| |
| − | </source>
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====Submitting a non-IB GPC job using openmpi=====
| |
| − | | |
| − | <source lang="sh">
| |
| − | #!/bin/bash
| |
| − | #PBS -l nodes=1:compute-eth:ppn=8,walltime=40:00:00,os=centos53computeA
| |
| − | #PBS -N 1
| |
| − | if [ "$PBS_ENVIRONMENT" != "PBS_INTERACTIVE" ]; then
| |
| − | if [ -n "$PBS_O_WORKDIR" ]; then
| |
| − | cd $PBS_O_WORKDIR
| |
| − | fi
| |
| − | fi
| |
| − | /scinet/gpc/mpi/openmpi/1.3.2-intel-v11.0-ofed/bin/mpirun
| |
| − | -mca btl_sm_num_fifos 7 -np $(wc -l $PBS_NODEFILE | gawk '{print $1}')
| |
| − | -mca btl self,sm -machinefile $PBS_NODEFILE
| |
| − | /scratch/cneale/exe/intel/gromacs-4.0.5/exec/bin/mdrun_openmpi -deffnm
| |
| − | pagp -nosum -dlb yes -npme 24 -cpt 120
| |
| − | ## To submit type: qsub this.sh
| |
| − | </source>
| |
| − | | |
| − | This is '''VERY IMPORTANT !!!'''
| |
| − | Please read the [[https://support.scinet.utoronto.ca/wiki/index.php/User_Tips#Running_single_node_MPI_jobs relevant user tips section]] for information that is essential for your single node (up to 8 core) MPI GROMACS jobs.
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 14 September 2009
| |
| − | | |
| − | =====Things still left to do for GROMACS=====
| |
| − | | |
| − | Intel has it's own fast fourier transform library, which we expect to yield improved performance over fftw.
| |
| − | We have not yet attempted such a compilation.
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 18 August 2009
| |
| − | =====GROMACS benchmarks on Scinet=====
| |
| − | | |
| − | This is a rudimentary list of scaling information.
| |
| − |
| |
| − | I have a 50K atom system running performance on GPC right now. On 56
| |
| − | cores connected with IB I am getting 55 ns/day. I set up 50 such
| |
| − | simulations, each with 2 proteins in a bilayer and I'm getting a total
| |
| − | of 5.5 us per day. I am using gromacs 4.0.5 and a 5
| |
| − | fs timestep by fixing the bond lengths and all angles involving
| |
| − | hydrogen.
| |
| − | | |
| − | I can get about 12 ns/day on 8 cores of the non-IB part of GPC -- also
| |
| − | excellent.
| |
| − | | |
| − | As for larger systems, My speedup over saw.sharcnet.ca for a 1e6 atom
| |
| − | system is only 1.2x running on 128 cores in single precision. Although saw.sharcnet.ca
| |
| − | is composed of xeons, they are running at 2.83 GHz (https://www.sharcnet.ca/my/systems/show/41), which is a
| |
| − | faster clock speed than the Scinet 2.5 GHz for Intel's next-generation X86-CPU architecture.
| |
| − | While GROMACS is generally not excellent for scaling up to or beyond 128 cores (even for large systems),
| |
| − | our benchmarking of this system on saw.sharcnet.ca indicated that it was running at about 65% efficiency.
| |
| − | Benchmarking was also done on Scinet for this system, but was not recorded as we were mostly tinkering with the
| |
| − | -npme option to mdrun in an attempt to optimize it. My recollection, though, is that the scaling was similar on scinet.
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 19 August 2009
| |
| − | =====Strong scaling for GROMACS on GPC=====
| |
| − | | |
| − | Requested, and on our list to complete, but not yet available in a complete chart form.
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 19 August 2009
| |
| − | =====Scientific studies being carried out using GROMACS on GPC=====
| |
| − | | |
| − | Requested, but not yet available
| |
| − | | |
| − | -- [[User:Cneale|cneale]] 19 August 2009
| |
| | ====LAMMPS==== | | ====LAMMPS==== |
| | ====NAMD==== | | ====NAMD==== |
Astrophysics
Athena (explicit, uniform grid MHD code)
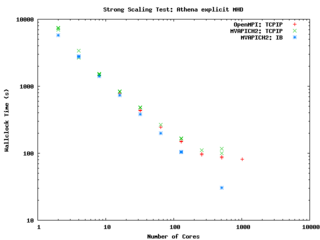
Athena scaling on GPC with OpenMPI and MVAPICH2 on GigE, and OpenMPI on InfiniBand
Athena is a straightforward C code which doesn't use a lot of libraries so it is pretty straightforward to build and compile on new machines.
It encapsulates its compiler flags, etc in an Makeoptions.in file which is then processed by configure. I've used the following additions to Makeoptions.in on TCS and GPC:
<source lang="sh">
ifeq ($(MACHINE),scinettcs)
CC = mpcc_r
LDR = mpcc_r
OPT = -O5 -q64 -qarch=pwr6 -qtune=pwr6 -qcache=auto -qlargepage -qstrict
MPIINC =
MPILIB =
CFLAGS = $(OPT)
LIB = -ldl -lm
else
ifeq ($(MACHINE),scinetgpc)
CC = mpicc
LDR = mpicc
OPT = -O3
MPIINC =
MPILIB =
CFLAGS = $(OPT)
LIB = -lm
else
...
endif
endif
</source>
It performs quite well on the GPC, scaling extremely well even on a strong scaling test out to about 256 cores (32 nodes) on Gigabit ethernet, and performing beautifully on InfiniBand out to 512 cores (64 nodes).
-- ljdursi 19:20, 13 August 2009 (UTC)
FLASH3 (Adaptive Mesh reactive hydrodynamics; explict hydro/MHD)
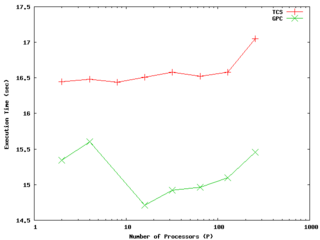
Weak scaling test of the 2d sod problem on both the GPC and TCS. The results are actually somewhat faster on the GPC; in both cases (weak) scaling is very good out at least to 256 cores
FLASH encapsulates its machine-dependant information in the FLASH3/sites directory. For the GPC, you'll have to
module load intel
module load openmpi
module load hdf5/183-v16-openmpi
and with that, the following file (sites/scinetgpc/Makefile.h) works for me:
<source lang="sh">
- Must do module load hdf5/183-v16-openmpi
HDF5_PATH = ${SCINET_HDF5_BASE}
ZLIB_PATH = /usr/local
- ----------------------------------------------------------------------------
- Compiler and linker commands
- We use the f90 compiler as the linker, so some C libraries may explicitly
- need to be added into the link line.
- ----------------------------------------------------------------------------
- modules will put the right mpi in our path
FCOMP = mpif77
CCOMP = mpicc
CPPCOMP = mpiCC
LINK = mpif77
- ----------------------------------------------------------------------------
- Compilation flags
- Three sets of compilation/linking flags are defined: one for optimized
- code, one for testing, and one for debugging. The default is to use the
- _OPT version. Specifying -debug to setup will pick the _DEBUG version,
- these should enable bounds checking. Specifying -test is used for
- flash_test, and is set for quick code generation, and (sometimes)
- profiling. The Makefile generated by setup will assign the generic token
- (ex. FFLAGS) to the proper set of flags (ex. FFLAGS_OPT).
- ----------------------------------------------------------------------------
FFLAGS_OPT = -c -r8 -i4 -O3 -xSSE4.2
FFLAGS_DEBUG = -c -g -r8 -i4 -O0
FFLAGS_TEST = -c -r8 -i4
- if we are using HDF5, we need to specify the path to the include files
CFLAGS_HDF5 = -I${HDF5_PATH}/include
CFLAGS_OPT = -c -O3 -xSSE4.2
CFLAGS_TEST = -c -O2
CFLAGS_DEBUG = -c -g
MDEFS =
.SUFFIXES: .o .c .f .F .h .fh .F90 .f90
- ----------------------------------------------------------------------------
- Linker flags
- There is a seperate version of the linker flags for each of the _OPT,
- _DEBUG, and _TEST cases.
- ----------------------------------------------------------------------------
LFLAGS_OPT = -o
LFLAGS_TEST = -o
LFLAGS_DEBUG = -g -o
MACHOBJ =
MV = mv -f
AR = ar -r
RM = rm -f
CD = cd
RL = ranlib
ECHO = echo
</source>
-- ljdursi 22:11, 13 August 2009 (UTC)
Aeronautics
Chemistry
GAMESS (US)
The GAMESS version January 12, 2009 R3 was built using the Intel v11.1 compilers and v3.2.2 MPI library, according to the instructions in http://software.intel.com/en-us/articles/building-gamess-with-intel-compilers-intel-mkl-and-intel-mpi-on-linux/
The required build scripts - comp, compall, lked - and run script - rungms - were modified to account for our own installation. In order to build GAMESS one first must ensure that the intel and intelmpi modules are loaded ("module load intel intelmpi"). This applies to running GAMESS as well.
The module "gamess" must also be loaded in order to run GAMESS ("module load gamess").
The modified scripts are in the file /scinet/gpc/src/gamess-on-scinet.tar.gz
Running GAMESS
- Make sure the directory /scratch/$USER/gamess-scratch exists.
- Make sure the modules: intel, intelmpi, gamess are loaded (in your .bashrc: "module load intel intelmpi gamess").
- Create a torque script to run GAMESS. Here is an example:
- The GAMESS executable is in $SCINET_GAMESS_BASE/gamess.00.x
- The rungms script is in $SCINET_GAMESS_BASE/rungms
- The rungms script takes 4 arguments: input file, executable number, number of compute processes, processors per node
For example, in order to run with the input file /scratch/$USER/gamesstest01, on 8 cpus, and the default version (00)
of the executable on a machine with 8 cores:
rungms /scratch/$USER/gamesstest01 00 8 8
-- dgruner 15 September 2009
Climate Modelling
Medicine/Bio
High Energy Physics
Structural Biology
Molecular simulation of proteins, lipids, carbohydrates, and other biologically relevant molecules.
Molecular Dynamics (MD) simulation
DESMOND
GROMACS
Please refer to the GROMACS page
LAMMPS
NAMD
NAMD is one of the better scaling MD packages out there. With sufficiently large systems, it is able to scale to hundreds or thousands of cores on Scinet. Below are details for compiling and running NAMD on Scinet.
More information regarding performance and different compile options coming soon...
Compiling NAMD for GPC
Ensure the proper compiler/mpi modules are loaded.
<source lang="sh">
module load intel
module load openmpi/1.3.3-intel-v11.0-ofed
</source>
Compile Charm++ and NAMD
<source lang="sh">
- Unpack source files and get required support libraries
tar -xzf NAMD_2.7b1_Source.tar.gz
cd NAMD_2.7b1_Source
tar -xf charm-6.1.tar
wget http://www.ks.uiuc.edu/Research/namd/libraries/fftw-linux-x86_64.tar.gz
wget http://www.ks.uiuc.edu/Research/namd/libraries/tcl-linux-x86_64.tar.gz
tar -xzf fftw-linux-x86_64.tar.gz; mv linux-x86_64 fftw
tar -xzf tcl-linux-x86_64.tar.gz; mv linux-x86_64 tcl
- Compile Charm++
cd charm-6.1
./build charm++ mpi-linux-x86_64 icc --basedir /scinet/gpc/mpi/openmpi/1.3.3-intel-v11.0-ofed/ --no-shared -O -DCMK_OPTIMIZE=1
cd ..
- Compile NAMD.
- Edit arch/Linux-x86_64-icc.arch and add "-lmpi" to the end of the CXXOPTS and COPTS line.
- Make a builds directory if you want different versions of NAMD compiled at the same time.
mkdir builds
./config builds/Linux-x86_64-icc --charm-arch mpi-linux-x86_64-icc
cd builds/Linux-x86_64-icc/
make -j4 namd2 # Adjust value of j as desired to specify number of simultaneous make targets.
</source>
--Cmadill 16:18, 27 August 2009 (UTC)
Monte Carlo (MC) simulation

